General
- Premature (before birth) fusion (ossification) or failure of separation of sutures
- Normal Skull growth
Pathophysiology
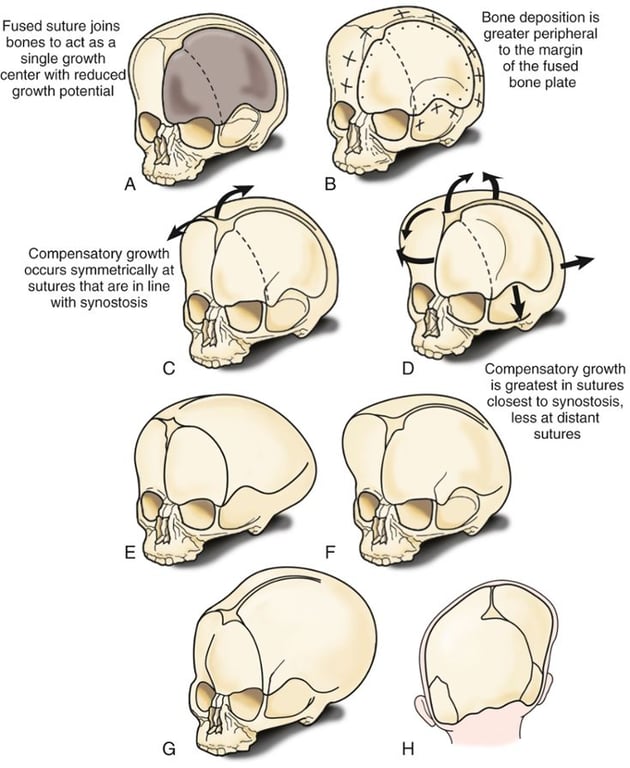
- Virchow’s law: if a suture prematurely fuses, growth is arrested perpendicular to the suture and is increased parallel to it.
- When one suture is closed to growth other sutures will try to compensate to allow for brain growth
- Skull growth is arrested in the direction perpendicular to the fused suture and expanded at the sites of unaffected sutures
Types
Syndromic vs Non syndromic/Sporadic
Syndromic (15% cases)
- Severity
- Mild cases
- No functional complications
- Management
- Conservative
- Surgical treatment
- Indication
- Cosmesis
- Defer definitive surgery until growth is complete
- Reducing pschological impact of deformity
- Severe cases
- Management
- Surgery
- indication
- Aim to preserve function by managing complication associated with
- Airway
- ICP
- Exorbitism
- Failure to thrive
- Cosmesis
- Defer definitive surgery until growth is complete
- Reducing psychological impact of deformity
- Associated tumour types
- Craniopharyngiomas (6-9%)
- Optic Pathway Hypothalamic Astrocytomas (2-7%)
- Germinomas (3%)
- Pituitary Adenomas (1-2%)
- Dermoids / Epidermoids
- Other:
- Meningiomas
- Haemangiomas
- Ependymomas
- Schwannomas
- Cavernomas
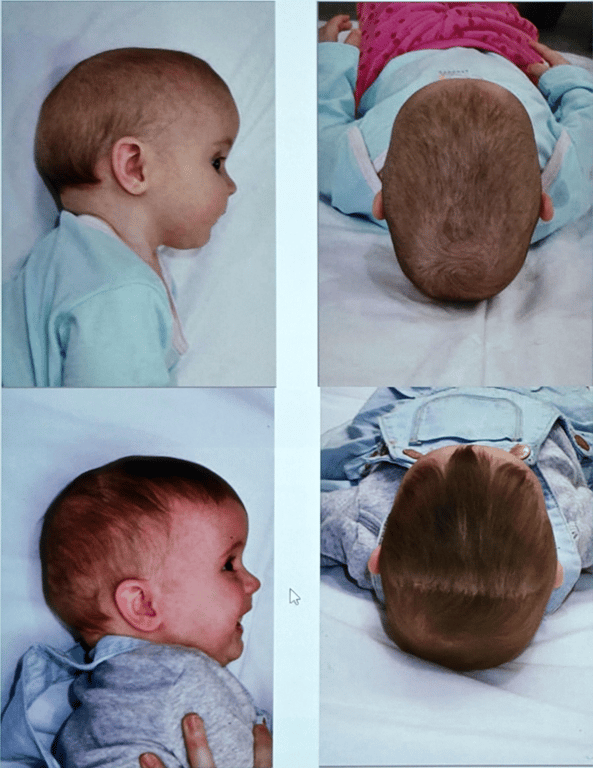
Phenotypic Features
Images
Genetic Mutations
Surgical indication
Muenke’s syndrome
- Unicoronal or bicoronal craniosynostosis
- Brachydactyly
- Thimble-like middle phalanges
- Coned epiphyses
- Carpal and tarsal fusions
- Sensorineural hearing loss
- Developmental delay
- Learning difficulties
- Seizures
- FGFR3
- Autosomal dominant.
- Caused by a point mutation in Pr0250Arg in the Ig 11-111 linker region of the FGFR3 gene on chromosome 4p16.3
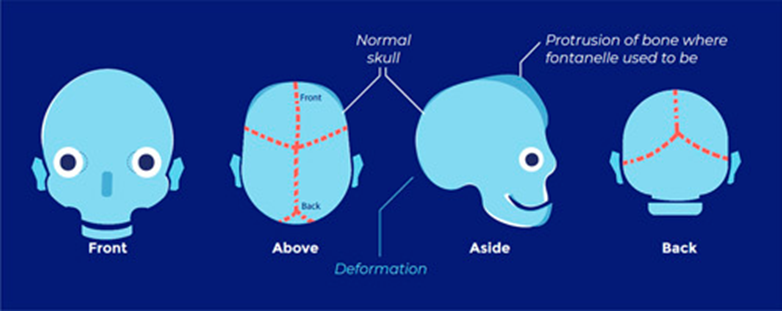
Abnormal skull shape
Low risk of raised ICP
Low risk of raised ICP
Saethre-Chotzen syndrome
- Manifestations very variable → need genetic testing
- Coronal craniosynostosis with limb abnormalities (syndactyly of the second and third digits, bifid hallux)
- Facial abnormalities (facial asymmetry, low frontal hairline, ptosis, and small ears with prominent ear crura)
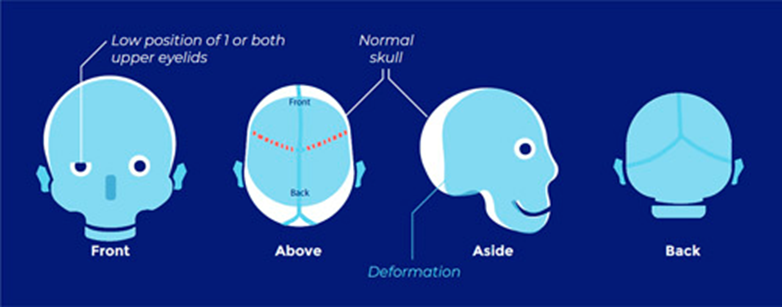
- TWIST1 Chr 7
- Autosomal dominant with complete penetrance and variable expressivity.
- Caused by loss-of-function mutations in TWIST (missense, nonsense, deletions, insertions, and duplications). Report of Q289P mutation in FGFR2
Abnormal skull shape Risk of ICP.
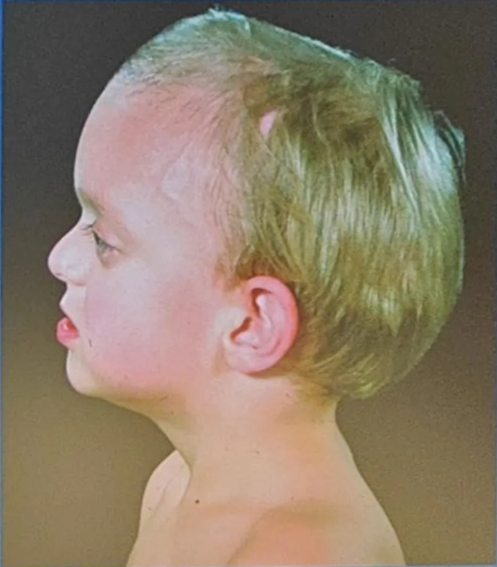
Crouzon’s syndrome
- Most common
- Classical triad
- Coronal synostosis
- Midfacial hypoplasia
- Exophthalmos
- Other features
- May also include involvement of other calvarial sutures
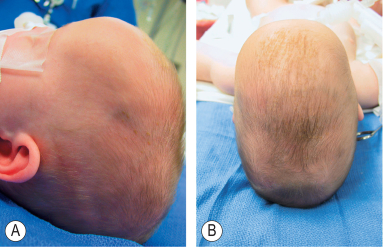
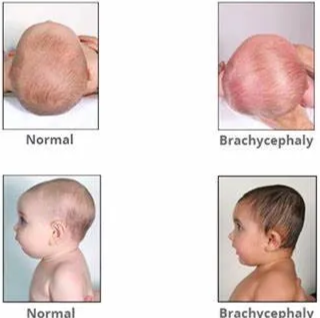
- Brachycephaly
- Hypertelorism
- Chiari I malformation
- Hydrocephalus
- Mental retardation
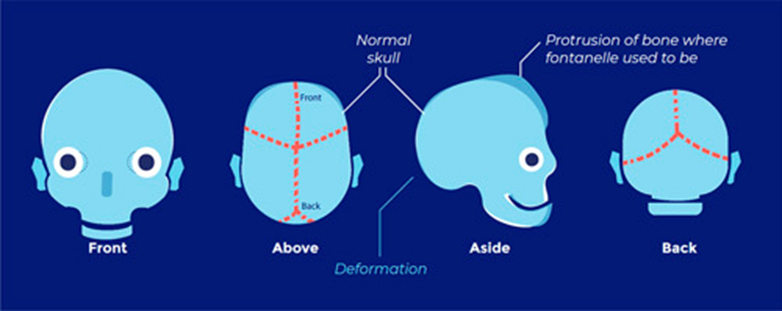
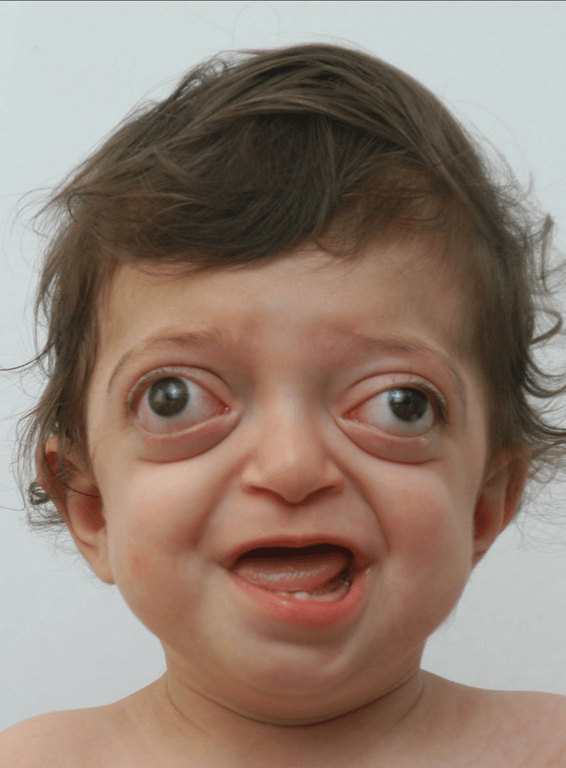
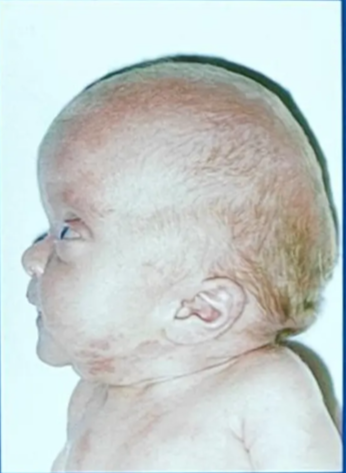
- FGFR2 Chr 10
- Autosomal dominant.
- Caused by numerous missense mutations in the Ig 111 domain of the FGFR2 gene (many involve the gain or loss of a cysteine residue)
Abnormal skull shape Risk of ICP.
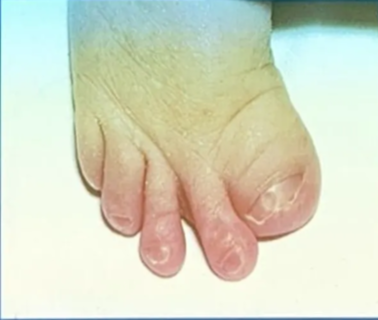
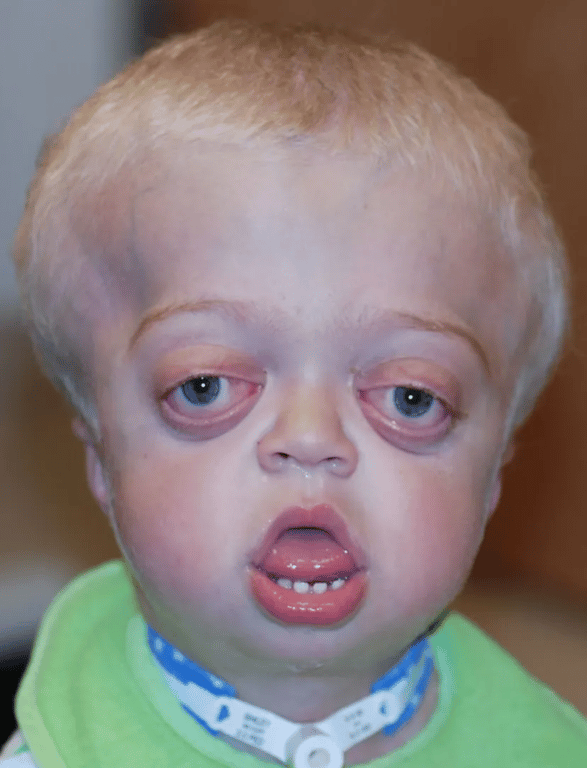
Pfeiffer’s syndrome
- Classical features
- brachycephaly
- membranous syndactyly of hands and feet with enlarged and deviated thumbs and great toes
- Types
- Type I classical Pfeiffer’s syndrome is a mild entity
- Types II and III are more severe, with early death.
- Other features
- Coronal synostosis with or without premature fusion of other calvarial sutures
- Cloverleaf skull
- Facial
- maxillary hypoplasia
- small nose with a low nasal bridge
- hypertelorism
- shallow orbits
- proptosis
- strabismus
- limb malformation
- Radiohumeral synostosis
- Broad fingers and toes
- partial syndactyly of the fingers and toes

- FGFR1, FGFR2
- Type I: autosomal dominant.
- Caused by FGFR1mutations, including Pr0252Arg in the Ig II-III linker region on chromosome 8p11.2-p11.
- Can also be caused by mutations in FGFR2 and be associated with more severe phenotypic expression.
- Types II and III: sporadic inheritance.
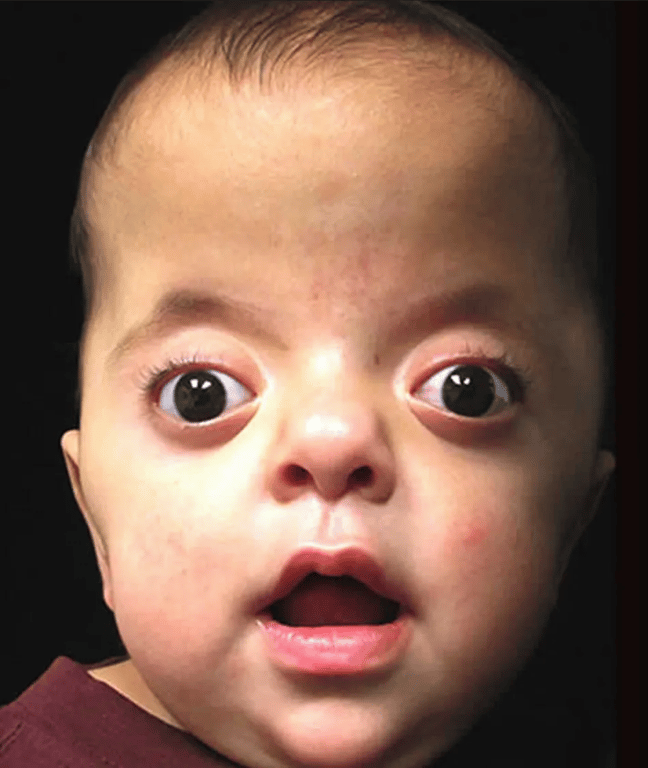
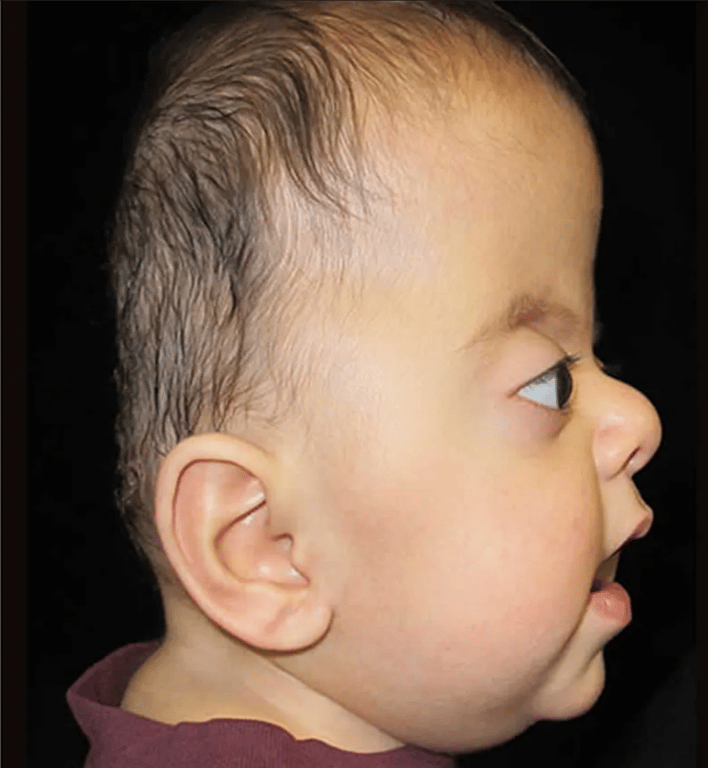
Apert’s syndrome (Acrocephalosyndactyly)
- 2nd most common
- Features
- Bicoronal synostosis
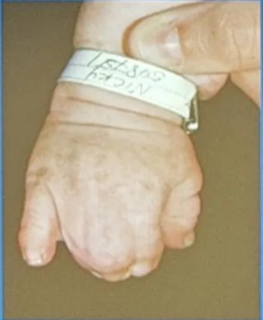
- Severe polysyndactyly in the fingers and toes
- Symphalangism (fusion of the phalanges)
- Radiohumeral fusion
- Mental retardation (IQ can be normal or mildly reduced)
- Antimongoloid slanted eyes
- Maxillary hypoplasia
- Cheerful effect
- Vs Crouzon's at the faciocranial level
- is the presence of hypertelorism and an open bite, in which the anterior part of the maxillary alveolar arch is higher than the posterior part.
- The face and the forehead are also abnormally wide, and the anterior fontanelle is widely open during the first months of life.
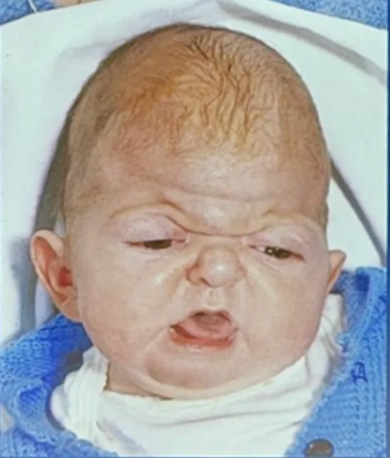
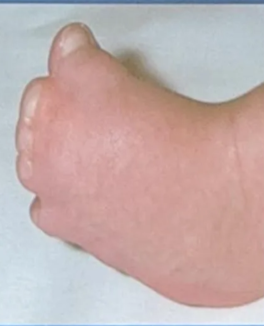
- FGFR2
- Autosomal dominant.
- Caused by a number of different mutations in FGFR2 on chromosome 10q26, including two missense mutations (Ser252Trp, Pr0253Arg) and two Alu insertions
Abnormal skull shape Risk of ICP.
Jackson-Weiss syndrome
- Like Crouzon's but with added enlarged great toes and tarsometatarsic fusion.
- Craniosynostosis, with broad toes and a medially deviated great toe, second and third toe syndactyly, tarsal-metatarsal fusion, broad and short metatarsals and proximal phalanges, midfacial hypoplasia, hypertelorism, proptosis, and normal intelligence
- FGFR2
- Autosomal dominant.
- Caused by mutation A344G in the highly conserved Ig IIIc domain of FGFR2,as well as two nucleotide missense mutations that result in Cys342Ser and Cys342Arg
Non-syndromic (85% cases)
- NOT 'GENETIC' (at least not germline...)
- but probably non syndromic one has not been diagnosed yet
- Predominantly appearance issue
- Aesthetic >> Functional
- Low chance of developing raised intracranial pressure
- Sporadic >> familial
- Usually "fixed" with one operation
Singe suture vs Multi-suture
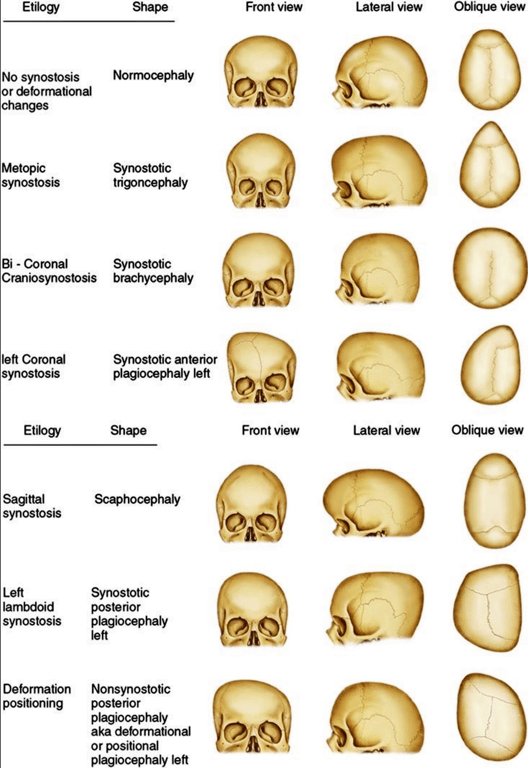
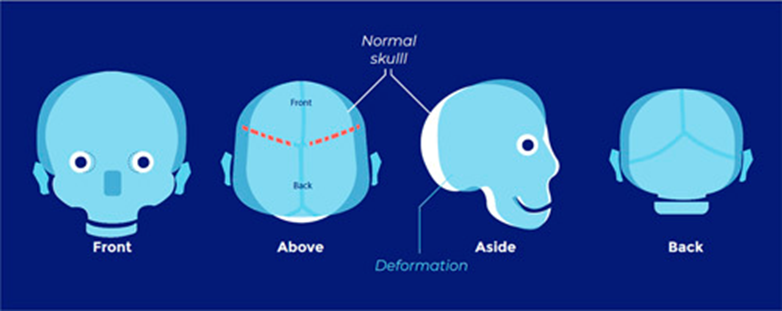
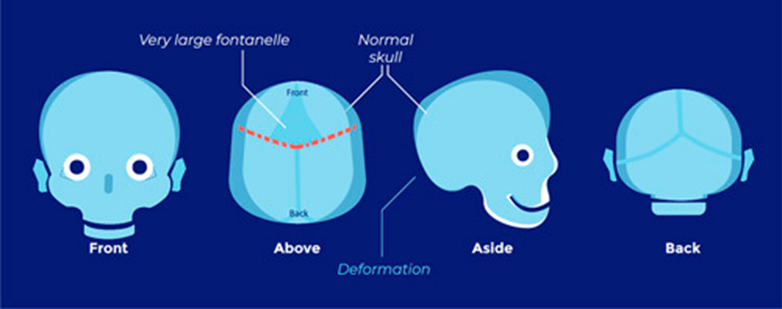
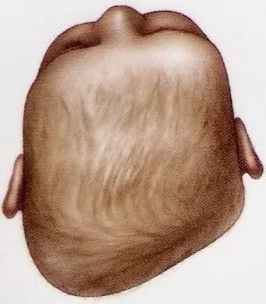
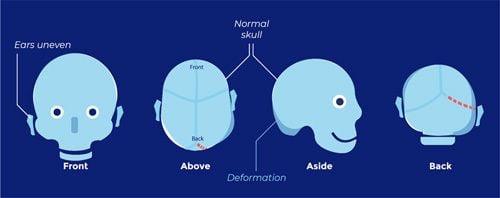
Positional plagiocephaly
- Aka Plagiocephaly without craniosynostosis
- Asymmetrical distortion (flattening of one side) of the skull.
- Mech
- Not due to craniosynostosis
- Due to
- Decreased mobility: patients who constantly lie supine with the head to the same side, e.g. cerebral palsy, mental retardation, prematurity, chronic illness
- Abnormal postures:
- Congenital torticollis
- Congenital disorders of the cervical spine
- Intentional positioning:
- due to the recommendation in 1992 to place newborns in a supine sleeping position to reduce the risk of sudden infant death syndrome (SIDS), sometimes with a foam wedge to tilt the child to one side to reduce the risk of aspiration
- Intrauterine aetiologies:
- Intrauterine crowding (e.g. from multiparous births or large foetal size)
- Uterine anomalies
- Differentiating between positional vs lambdoid synostosis
- When born round and normal head then change it is positional
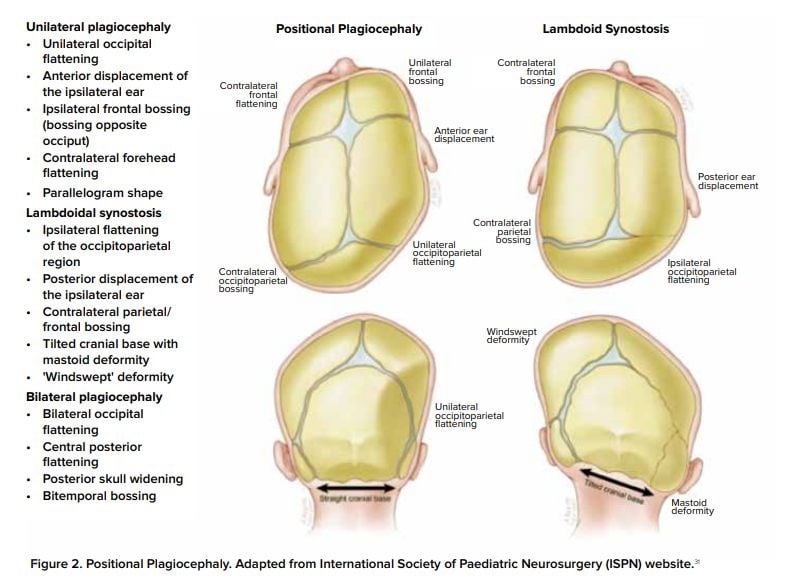
Condition | Head Shape | Ear Displacement | Frontal/Occipital Bossing |
Posterior Deformational Plagiocephaly | Parallelogram shaped head | Anterior displacement of ipsilateral ear | Ipsilateral frontal bossing |
Unilateral Lambdoid Synostosis | Trapezoid shaped skull | Posterior displacement of ipsilateral ear | ㅤ |
- Severity
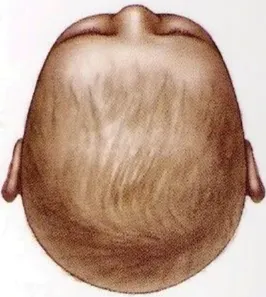
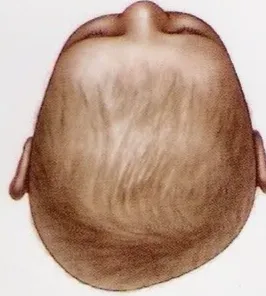
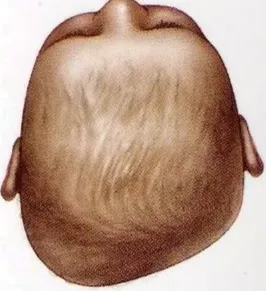
- Management
- Will improve over time
- Helmet therapy
- at 6-7 months of age
- Indication
- Moderate to severe cases unresponsive to repositioning
- Cases associated with torticollis

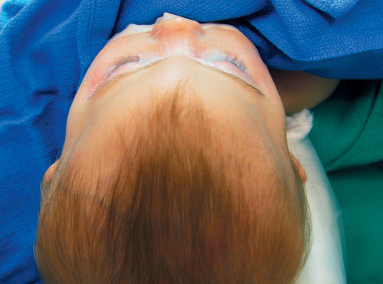
Sagittal craniosynostosis (scaphocephaly or dolichocephaly)
- Most common form of craniosynostosis (60%)
- Occurs at a rate of 1/5000 children
- male-to-female ratio of 3.5 : 1.62
- Have higher chance of language problems
- No evidence surgery improves language
- Features
- Sagittal ridge
- Small anterior fontanelle
- Bossed forehead
- Temporal pinching
- Occipital bullet
- Genetic mutations
- SMAD 6 mutation
- Missense mutations (S188L and S201Y) in the TWIST box have been identified in patients with isolated sagittal craniosynostosis.
- K526E mutation in the tyrosine kinase domain of the FGFR2 gene
- Susceptibility loci within BBS9 and near BMP2 (bone morphogenetic protein 2), which are known genes important for skeletal development.
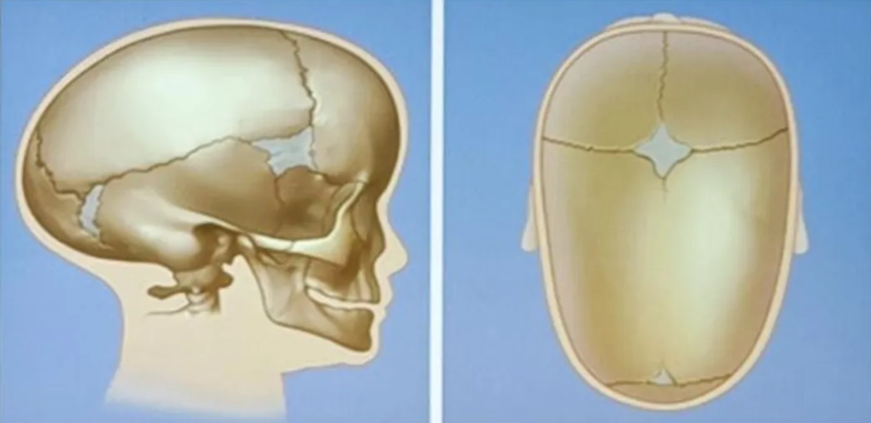
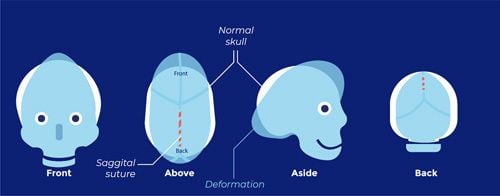
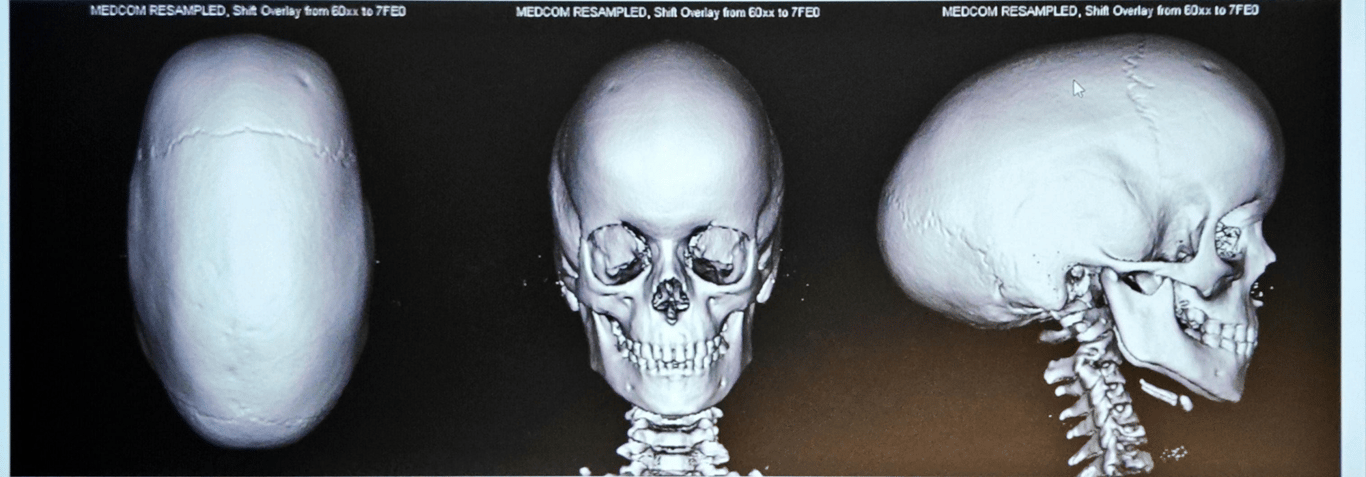
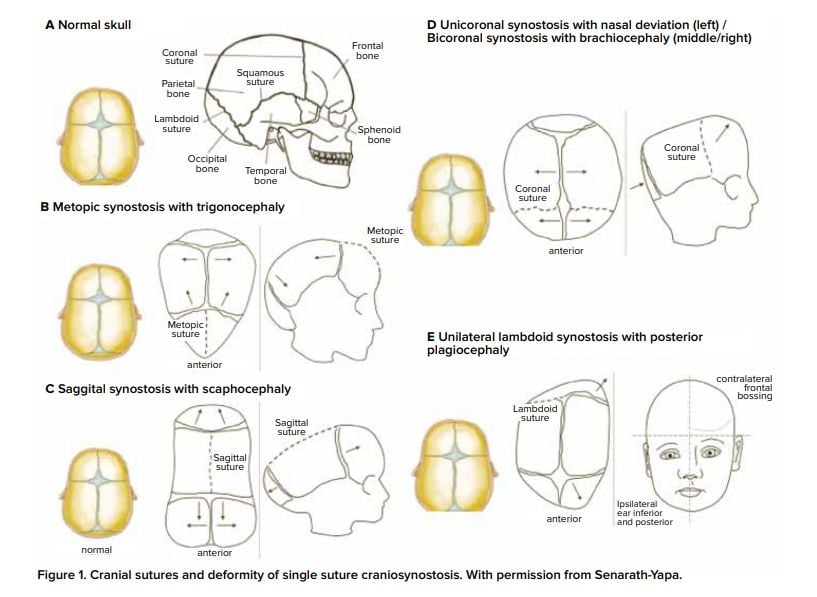
Coronal craniosynostosis
- Second most common sutural fusion (18% for unicoronal)
- Occurs at a rate of 1/10,000 children
- male-to-female ratio of 1 : 2.
- Because many cases of syndromic craniosynostosis, especially Muenke’s syndrome, may be associated with unicoronal synostosis, it is recommended that one should test for these mutations before diagnosing nonsyndromic coronal synostosis.
- Two types
- Unicoronal
- 20% have genetic disorder
- The side of the fused defect follows the root of the nose
- I.e. Left sided root of nose = left sided unicoronal suture craniosynostosis
- Can lead to shallow orbital vault: superior orbital rim is not protruding normally can lead to
- Harlequin eye deformity
- It is the shape seen on radiography of the orbit from a fused coronal suture found in a unilateral coronal synostosis. The elevation of the greater and lesser wings of the sphenoid ipsilateral to the fused suture give the eye this unusual shape.
- Strabismus is common (50-60%) due to mechanical effect on superior oblique, and anterior plagiocephaly is commoner on the right side (3:2).
- Bicoronal
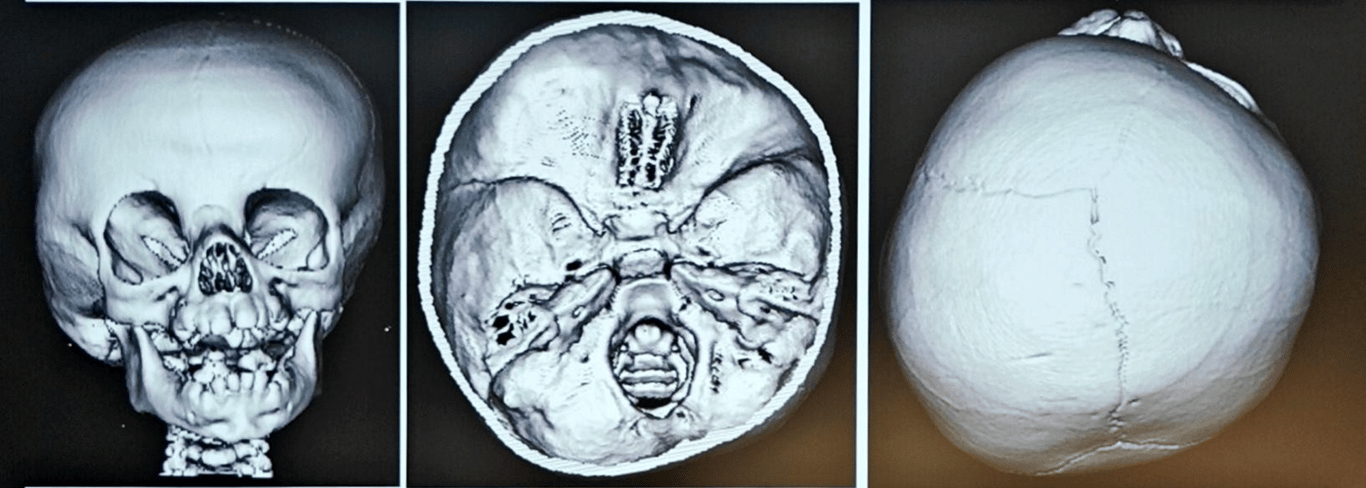
Metopic craniosynostosis
- Causes trigonocephaly
- Negligible rate of intracranial hypertension
- Third most common single-suture nonsyndromic craniosynostosis (25%)
- 1 out of 10,000 to 15,000 live births
- male-to-female ratio of 3.3 : 1.60
- Evidence that the incidence of metopic craniosynostosis may be increasing in the Northeastern United States and Europe, for unclear reasons.
- Aetiology of this phenotype is considered heterogeneous, with both genetic and environmental factors, such as prenatal head constraints playing a role
- Use of maternal valproate
- Genetic mutation
- RUNX2 possibly associated with it but unsure the prevalence of this mutation
- SMAD6

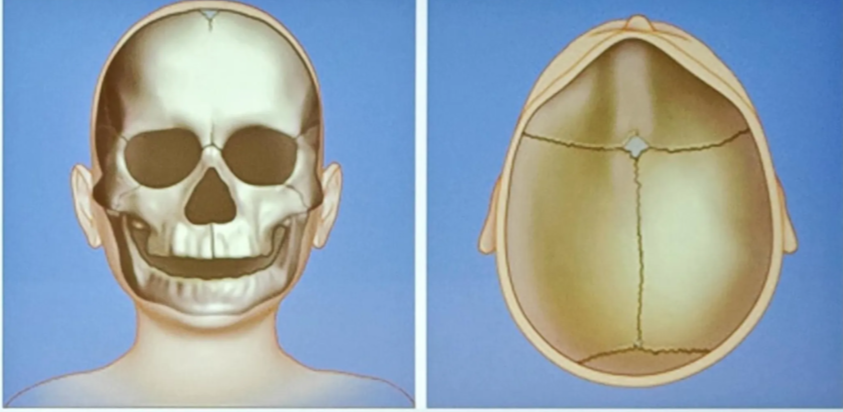
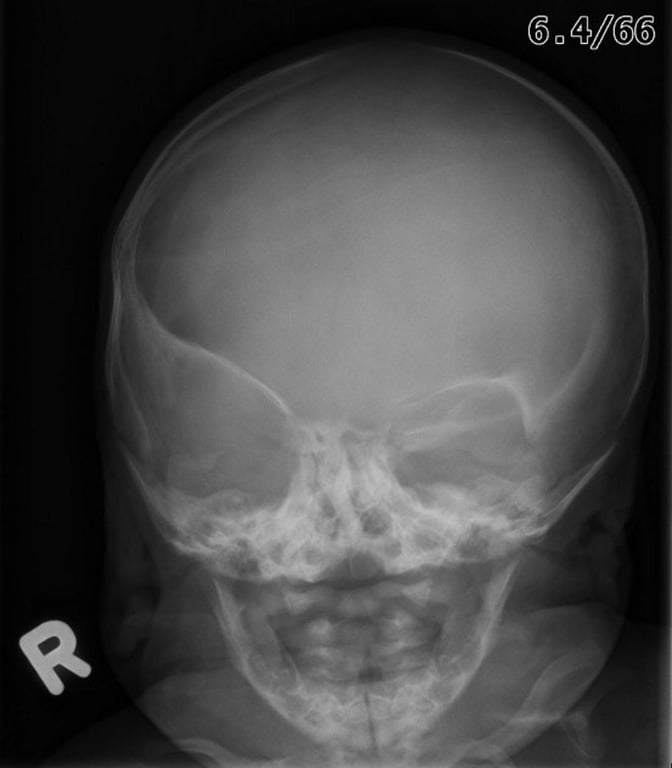
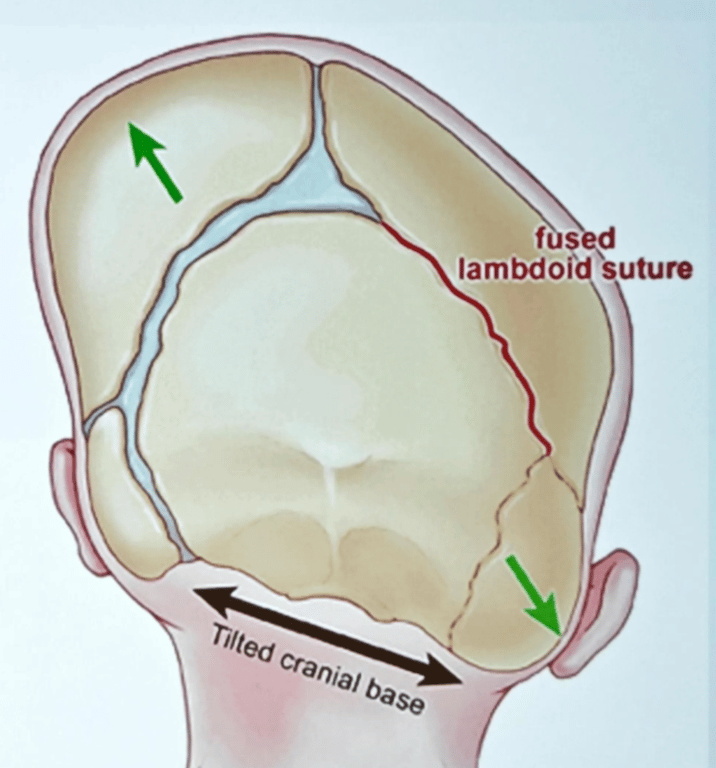
Lambdoid craniosynostosis
- Very rare (3%)
- May mimic positional plagiocephaly
- Can be associated with Chiari
Numbers
- Incidence: 0.6/1000 live births
Aetiology
- Primary
- is usually a prenatal deformity.
- Secondary
- Metabolic
- Rickets
- Hyperthyroidism
- Toxic
- Drugs (phenytoin, valproate, methotrexate)
- Hematologic
- Sickle cell
- Thalassemia
- Structural
- Lack of brain growth due to e.g. microcephaly, lissencephaly, micropolygyria
Presentation
Cosmetic deformity
- Some cases of “synostosis” are really deformities caused by positional flattening (lazy lambdoidal)
- To differentiate between lazy lambdoidal vs true craniosyntosis
- instruct parents to keep head off of flattened area and recheck patient in 6–8 weeks
- if it was positional, it should be improved, if it was CSO then it usually declares itself
- Physical examination
- palpation of a bony prominence over the suspected synostotic suture (exception: lambdoidal synostosis may produce a trough)
- gentle firm pressure with the thumbs fails to cause relative movement of the bones on either side of the suture
- Ways to describe the phenotypes (these phenotypes may or may not be due to a craniosynostosis)
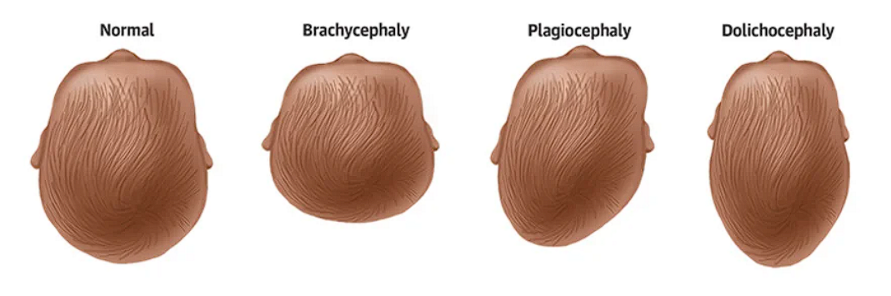
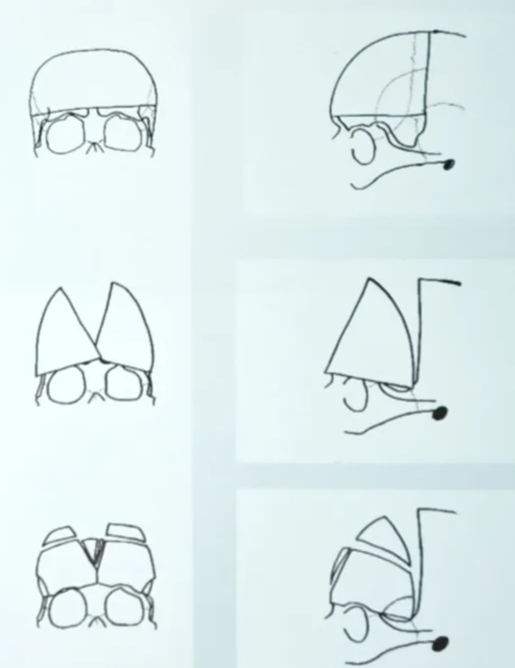
- Scaphocephaly (Dolichocephaly)
- Sagittal craniosynostosis
- Brachycephaly
- "brakhu" (short) and "cephalos" (head), which translates to "short head.
- Bilateral
- Coronal craniosynostosis
- Plagiocephaly
- plagios (oblique) and kephale (head), meaning distortion of the head
- Unilateral craniosynostosis
- Anterior plagiocephaly
- Unicoronal craniosynostosis
- Posterior plagiocephaly
- Lambdoid craniosynostosis
- Postural plagiocephaly
- Trigonocephaly
- Metopic craniosynostosis
- Kleeblattschadel/clover leaf deformity
- Due to premature closure of sagittal, coronal, and lambdoid sutures
- Look for features of
- Aperts
- Pfeiffer’s
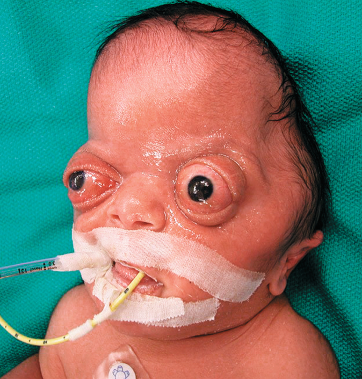
- Facial scoliosis
Multisutural CSO
- Brain growth impeded by unyielding skull
- Types of brain malformation
- Callosal agenesis
- Chronic tonsillar herniation
- Different from type 2 Chiari malformation
- Ventricular dilatation
- Present as (will require urgent decompression)
- Apnea/hypoapnea
- deleterious effect to brain growth
- breath-holding spells,
- vocal cord paralysis, and
- bulbar palsies
- Pathologically elevated ICP
- 11% of cases with a single stenotic suture
- radiographic signs (on plain skull X-ray or CT, see above)
- Two mechanism
- failure of calvarial growth (unlike the non-synostotic skull where increased ICP causes macrocrania in the newborn, here it is the synostosis that causes the increased ICP and lack of skull growth)
- altered venous outflow secondary to stenosis of the jugular foramina
- obstruction in CSF pathways as a result of distortion of cerebral structures (aqueduct and posterior fossa)
- The development of raised ICP is a progressive event, inasmuch as the incidence increases with age
- Present as
- papilledema --> optic atrophy and visual loss
- developmental delay/low IQ
- Elevated ICP correlates with poorer IQ --> tx raised ICP early before 1 year old
Learning difficulties
- can be due to:
- Genetic cause
- "Failure to thrive"
- Breathing difficulties
- Feeding difficulties
- Raised ICP
- Low expectations
- Physical disability
- Visual handicap
Raised ICP
- Due to
- HCP
- Craniocerebral disproportion
- Venous HTN
- Air way obstruction
- Raised PaCO2
Imaging
General
- Skull x-rays or an ultrasound of the skull are always done if there is a moderate suspicion of craniosynostosis.
- If there is a strong suspicion of craniosynostosis based on external features, a 3D-CT is immediately done for diagnostic purposes.
- Children with syndromic craniosynostosis are sometimes given additional MRI scans to assess other brain disorders and symptoms of increased intracranial pressure (ICP) before surgery.
plain skull X-rays:
- lack of normal lucency in center of suture.
- Some cases with normal X-ray appearance of the suture (even on CT) may be due to focal bony spicule formation
- beaten copper calvaria, sutural diastasis and erosion of the sella may be seen in cases of increased ICP
CT scan:
- To use CT or not
- CT scans performed early in infancy are associated with an increased likelihood of malignancy later in life
- Prospective multicenter study demonstrated that a correct diagnosis of craniosynostosis could be derived as accurately by simple physical examination as by CT scan
- To be used if
- A doubt in the diagnosis
- Surgery planning
- helps demonstrate cranial contour
- may show thickening and/or ridging at the site of synostosis
- will demonstrate hydrocephalus if present
- may show expansion of the frontal subarachnoid space
- three-dimensional CT may help better visualize abnormalities
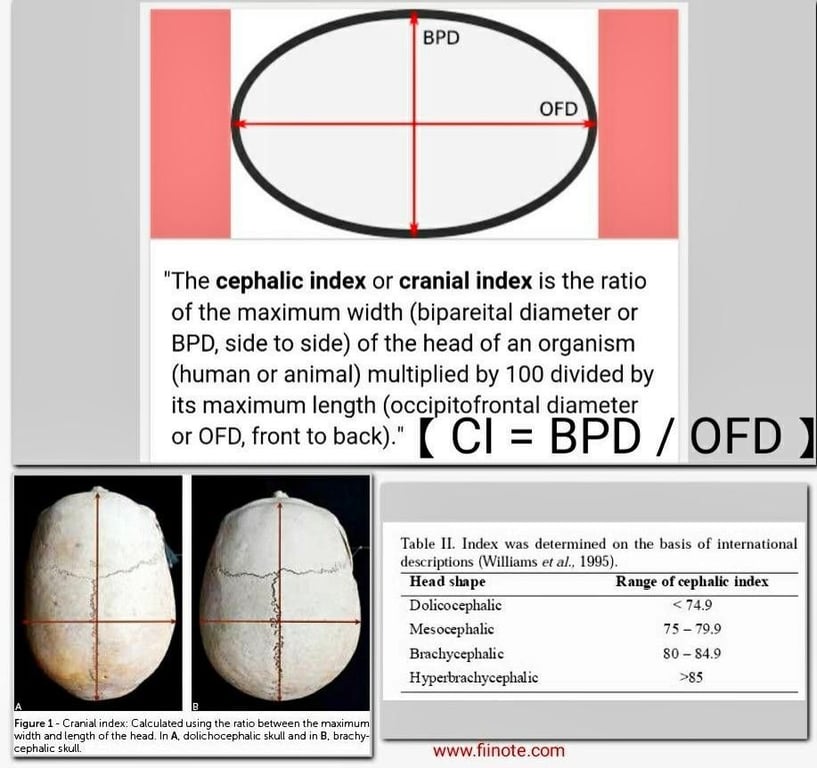
Measurements, such as occipito-frontal-circumference may not be abnormal even in the face of a deformed skull shape
Technetium bone scan
- indicated when there is dubiety of the presence of craniosynostosis
- there is little isotope uptake by any of the cranial sutures in the first weeks of life
- in prematurely closing sutures, increased activity compared to the other (normal) sutures will be demonstrated
- in completely closed sutures, no uptake will be demonstrated
MRI
- usually reserved for cases with associated intracranial abnormalities.
- Often not as helpful as CT
Management
Aim of treatment
- Cosmesis
- The external abnormality (with both esthetic and psychological consequences)
- Functional improvement (language)
- Correct strabismus
- Boston study
- Craniocerebral dysproportionism - HCP
- Preventing or limiting associated brain abnormalities
Conservative
- Children may be managed non-surgically for 3–6 months. → 15% will develop a significant cosmetic deformity
- Repositioning
- effective in ≈ 85% of cases.
- Patients are placed on the unaffected side or on the abdomen.
- Infants with occipital flattening from torticollis should have aggressive physical therapy and resolution should be observed within 3–6 months
- Trial of moulding helmets
Surgery
- General
- 20% pt will require
- ideal age for surgery is between 6 and 18 months
- Indication
- Raised ICP (Intracranial Pressure)
- Sort out airway
- Sleep studies
- Treat hydrocephalus
- ICP studies
- Vault expansion
- For craniocerebral disproportion
- If fail then shunts
- Cosmetic / Functional
- Surgical techniques
- Option:
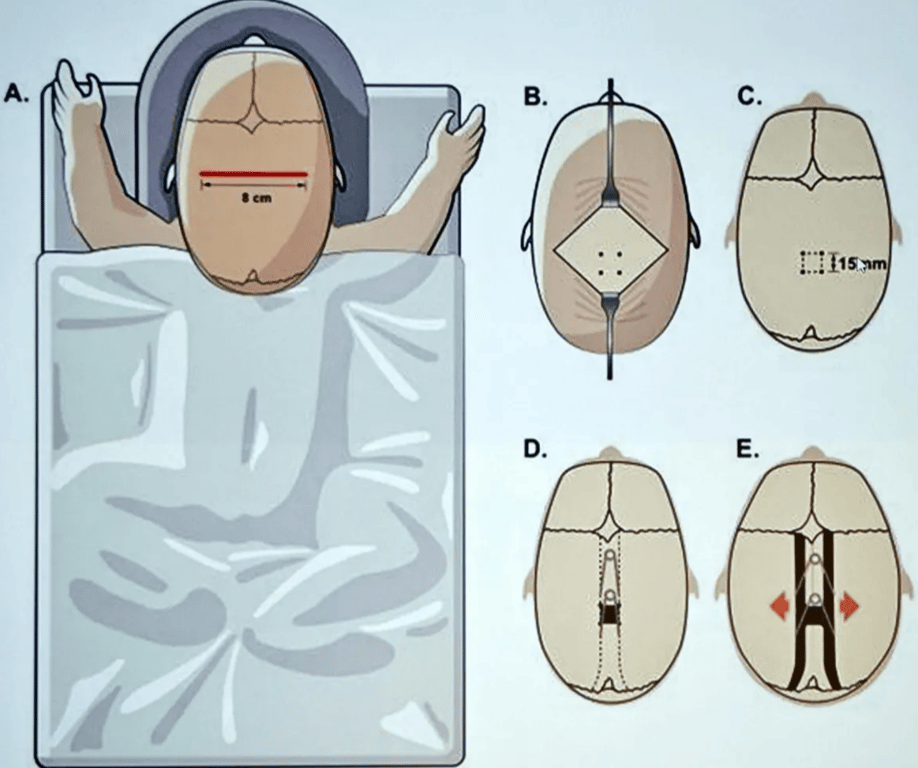
- Endoscopic suturectomy + helmet use
- Criteria
- done <3 months
- Pros
- less blood loss,
- fewer blood transfusions,
- shorter surgery duration and admission time
- a similar aesthetic result
- By preventing secondary changes to skull and midface
- Ophthalmic results after a minimally invasive procedure with coronal suture synostosis may also be better than with an open correction
- Cheaper
- Small scar
- Reduce strabismus
- Open skull correction
- Intraop prep
- Use tranexamic acid during surgery to limit blood loss.
- Consider collecting the patient’s blood during surgery (using a cell saver) and then returning it to limit the number of blood transfusions.
- Use fresh frozen plasma and/or fibrinogen as soon as signs of abnormal coagulation develop during surgery.
- Spring-assisted distraction
- in sagittal suture synostosis is probably less likely to lead to ICP in the years after surgery than an open skull correction
- Pros:
- Gradual process
- Minimal surgery and operative time
- Cons:
- Need another surgery to remove distraction device
- If left too long, metal device can migrate intracranially --> absorbable plates and screw
- Monobloc +/- distraction
- Bipartition +/- distraction
- Criteria
- Done >6 months
- The likelihood of developing ICP increases over the course of the first year of life (from 2.5% at 6 mo to 10% at 11 mo).
- Total calvarial remodelling
- Anterior remodelling (fronto-orbital remodelling)
- Improve forehead and orbital appearance
- Indicated
- Metopic, unicoronal
- Some bicoronal and sagittal patients
- Technique
- Bandeau (Marshac)
- Non-bandeau (Hayward)
- Midface surgery
- Reasons for It
- Functional
- Aesthetic
- Psychological
- Types of
- Monobloc
- Midfacial bipartition
- Le Fort III
- With or without RED frame
- Rigid external distraction
- Process of distraction osteogenesis divided into 4 phases:
- Osteotomy to divide bone to be lengthened
- Latent phase when callus allowed to form
- Period of active distraction at a rate of 1-2mm/24 hr
- Consolidation phase 6-8 weeks
- Minor
- Soft tissue realignments
- Dental
Minimal invasive surgery

- Risks
- blood loss
- Average blood loss for uncomplicated cases is 100–200 ml, and therefore transfusion is often required.
- Seizures
- Stroke
- CSF leak
- Infection
 Made with Bullet
Made with Bullet