General
- Definition: a group of rare bone disorders, within the family of sclerosing bone dysplasias, characterized by reduced osteoclastic bone resorption that results in a high bone mass.
- Derived from the Greek words for "bone" ("osteo") and "stone" ("petros")
Numbers
- Incidence:
- 1:200,000 for autosomal-recessive osteopetrosis
- 1:20,000 for autosomal-dominant osteopetrosis
Subtype
- “malignant” autosomal recessive infantile form
- Fatal first year of life
- Osteopetrosis has for decades been categorized descriptively by its clinical severity and inheritance pattern into a , a “benign” adult autosomal-dominant form (which can be severe), and an intermediate form
Pathophysiology
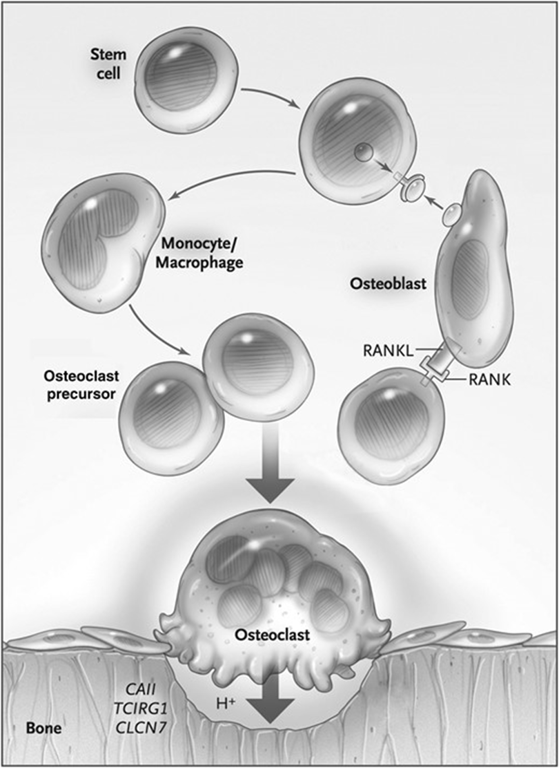
- Molecules affecting osteoclast differentiation and function.
- The binding of receptor activator of nuclear factor κ-B (RANK) ligand (RANKL) produced by osteoblasts to RANK on osteoclast precursors induces osteoclast differentiation;
- in animals with defects in these genes, no osteoclasts are observed.
- In disorders of acidification—there are no primary defects in differentiation reduced osteoclastic bone resorption that results in a high bone mass.
- including defects in carbonic anhydrase II (CAII), the osteoclast-specific proton pump (encoded by TCIRG1, also termed ATP6i), and the gene encoding a chloride channel (CLCN7)
- Normal bone resorption by Osteoclasts:
- Differentiation from granulocyte-macrophage precursors → osteoclast
- Osteoclast attachment to bone
- Creation of a ruffled border (resorptive surface)
- Acidification of bone to dissolve and digest the mineral matrix
- Generation of protons by carbonic anhydrase II (CAII)
- Transport of protons through the ruffled border by a vacuolar proton pump
- Maintenance of electroneutrality in the ruffled border by chloride exchange
- Defects in Osteopetrosis:
- Loss-of-function mutations impairing the acidification process through trafficking and/or fusion of lysosome-related organelles to the ruffled border
- eg of mutations:
- Osteoclast vacuolar proton pump (TCIRG1, MIM 604592)
- H+-Cl− exchange transporter 7 (CLCN7, MIM 602727)
- Account for nearly 70% of autosomal-recessive osteopetrosis
- CAII enzyme (MIM 611492)
- Stabilizing β-subunit of the H+-Cl− exchange transporter 7 (OSTM1, MIM 607649)
- Lysosome-associated protein involved in vesicular trafficking (PLEKHM1, MIM 611466)
- Osteoclast-Poor Forms of Autosomal Recessive Osteopetrosis:
- Mutations in tumor necrosis factor ligand superfamily member 11 (TNFSF11, MIM 602642)
- Tumor necrosis factor receptor superfamily member 11A (TNFRSF11A, MIM 603499)
- Interference with receptor activator of nuclear factor κ-B receptor and ligand signaling pathway
- Impact on osteoblast activation of osteoclastogenesis
- Due to poor bone reabsorption and net bone formation → Rather than conferring strength, the overly dense bone architecture forms a structural brittle bone that predisposes to fracture → give rise to skeletal deformity and dental abnormalities
- Can interfere with
- Mineral homeostasis expansion of bone into marrow cavities
- Manifest as profound anaemia, bleeding, frequent infections, and hepatosplenomegaly from extramedullary hematopoiesis.
- Cranial nerve foramina
- The latter can lead to blindness, deafness, and nerve palsies
flowchart LR A[Poor bone resorption and<br>net bone formation] B[Overly dense<br>bone architecture] C[Structural brittle bone] D[Skeletal deformity<br>and dental abnormalities] E[Interference with<br>mineral homeostasis] F[Expansion of bone<br>into marrow cavities] G[Profound anaemia, bleeding,<br>frequent infections,<br>hepatosplenomegaly from<br>extramedullary<br>hematopoiesis] H[Interference with cranial<br>nerve foramina] I[Blindness, deafness,<br>and nerve palsies] A --> B B --> C C --> D D --> E E --> F F --> G E --> H H --> I
Radiology
- Skeletal survey is sufficient to make the diagnosis.
- Because of the wide gamut of pathognomonic radiographic features of osteopetrosis,
- Parallel bands of dense bone can give the appearance of “bone-within-bone” or “endobones.”
- Seen most often in the
- pelvis,
- long bones,
- Club-shaped flaring of the metaphyses of long bones with cortical thinning (Erlenmeyer flask bone deformity) and transverse metaphyseal lucent bands reflect a failure of metaphyseal remodeling but are not exclusive to osteopetrosis
- phalanges,
- Vertebrae
- Vertebrae can also be uniformly dense or take on a “sandwich vertebrae” or “rugger-jersey” appearance (when a normal-appearing vertebral midbody is sandwiched between dense bands along the superior and inferior endplates).
- Evidence of new or healing fractures may be found, and skull changes can include calvarial and basilar thickening as well as poor sinus development.
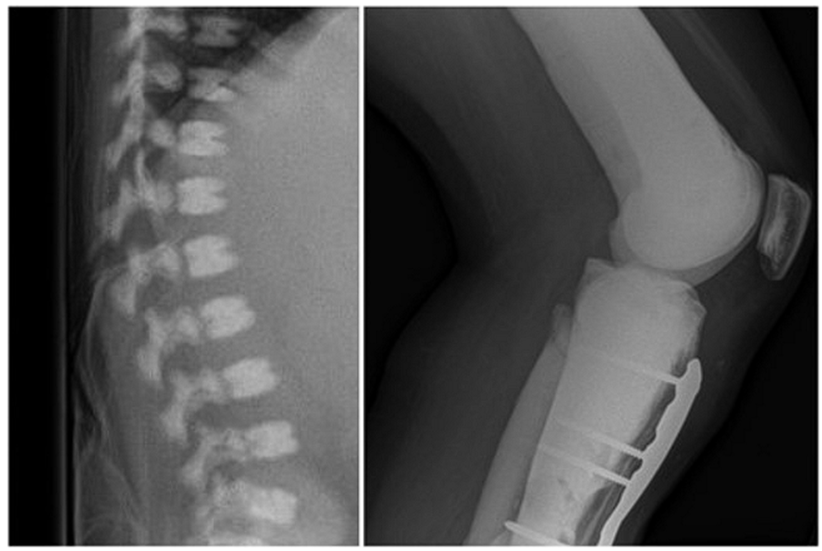
Right: Bone-within-bone appearance.
- Neurology and neurosurgery symptoms
- Most prevalent neurologic complications of osteopetrosis are the compressive neuropathies that stem from progressive constriction of cranial foramina and/or the failure of these foramina to enlarge proportionately with growth.
- Autosomal-recessive osteopetrosis
- The optic nerve most affected
- Optic nerve decompression (17, 25) and optic nerve sheath fenestration (44) in the appropriate clinical settings have been demonstrated to improve visual function and may better preserve visual acuity if performed early (45), although it is important to note that such positive outcomes are not universal (28, 46–48) and can be difficult to achieve in very young patients.
- autosomal-dominant CLCN7-deficient osteopetrosis
- most frequent cranial nerve deficit in is the facial nerve followed by the acoustic nerve, although any of cranial nerves I through VIII can be involved
- Many of the cranial palsies can be subclinical (41) and are detected only on careful evaluation or after they have progressed.
- Successful results have also been reported to follow decompression of the facial and acoustic nerves
- Less commonly reported are the consequences of calvarial thickening reducing cranial capacity, which can include increased ICP, posterior fossa crowding, cerebellar tonsillar herniation, and hydrocephalus
- Cerebrovascular stenosis and occlusion
- Craniosynostosis
- Arnold–Chiari I malformation
- Neurologic disease is associated with some specific genetic mutations.
- Developmental delay or regression, unexplained seizures, and neuroradiologic findings should alert to the possibility of an OSTM1 mutation.
- The cognitive defects of CAII deficiency are more variable, but the calcifications involving the basal ganglia, thalami, and/or gray-matter junction are almost always present
Other associated complication
Subspecialty | Complication |
Endocrinology | Osteopetrorickets Hypocalcemia |
Ophthalmology | Papilledema Ptosis Strabismus Paralysis of extraocular muscles Optic nerve atrophy Exophthalmos Nystagmus Retinal degeneration Tearing (from nasolacrimal duct obstruction) |
Dentistry | Delay/failure of tooth eruption Malformed crowns/roots Periodontal ligament defects Odontoma Tooth agenesis Enamel hypoplasia Tooth decay/caries Thickened lamina dura Osteomyelitis (most frequently of the mandible) |
Orthopedics | Skeletal deformities Scoliosis Spondylolisthesis Fractures (particularly of the long bones) Delayed union/nonunion Degenerative arthritis Spondylolysis |
Neurology/Neurosurgery | Compressive cranial neuropathies (often optic and facial nerves, but can involve any of cranial nerves I-VIII) Increased intracranial pressure Craniosynostosis Arnold-Chiari I malformation Neuromuscular scoliosis Developmental delay/regression, seizures (OSTM1 mutation) Calcifications of the basal ganglia, thalami (CAII deficiency) Hydrocephalus Cerebrovascular stenosis/occlusion Acquired encephalocele |
Otolaryngology | Conductive hearing loss Recurrent otitis media Chronic congestion (poorly pneumatized sinuses) Rhinorrhea Choanal atresia Rhinosinusitis Obstructive sleep apnea |
Hematology | Thrombocytopenia with bleeding Anemia Leukopenia with frequent infections Hepatosplenomegaly Transfusion dependence |
Nephrology | Renal tubular acidosis, nephrocalcinosis, and nephrolithiasis (CAII deficiency) |
Screening and Monitoring for patients with Osteopetrosis
Intervention | Timing |
Serum calcium | At diagnosis and every 6–12 months (every 3 months during calcitriol therapy) |
Serum phosphorus | At diagnosis and every 6–12 months (every 3 months during calcitriol therapy) |
Serum creatinine | At diagnosis (every 3 months during calcitriol therapy) |
25(OH)D | At diagnosis and every 6–12 months |
Parathyroid hormone | At diagnosis and every 6–12 months |
Complete blood count with differential | At diagnosis and every 6–12 months |
BB isozyme of creatine kinase | At diagnosis |
Lactate dehydrogenase | At diagnosis |
MRI to evaluate optic nerves | At diagnosis and as clinically indicated |
Urinary calcium/creatinine ratio (random or 24 hours) | At initiation of calcitriol and every 3 months during calcitriol therapy |
Renal ultrasonography | At diagnosis and every 12 months in patients with CAII mutations (every 12 months during calcitriol therapy) |
 Made with Bullet
Made with Bullet