Syndromic (15% cases)
- Severity
- Mild cases
- No functional complications
- Management
- Conservative
- Surgical treatment
- Indication
- Cosmesis
- Defer definitive surgery until growth is complete
- Reducing pschological impact of deformity
- Severe cases
- Management
- Surgery
- indication
- Aim to preserve function by managing complication associated with
- Airway
- ICP
- Exorbitism
- Failure to thrive
- Cosmesis
- Defer definitive surgery until growth is complete
- Reducing psychological impact of deformity
- Associated tumour types
- Craniopharyngiomas (6-9%)
- Optic Pathway Hypothalamic Astrocytomas (2-7%)
- Germinomas (3%)
- Pituitary Adenomas (1-2%)
- Dermoids / Epidermoids
- Other:
- Meningiomas
- Haemangiomas
- Ependymomas
- Schwannomas
- Cavernomas
Phenotypic Features
Images
Genetic Mutations
Surgical indication
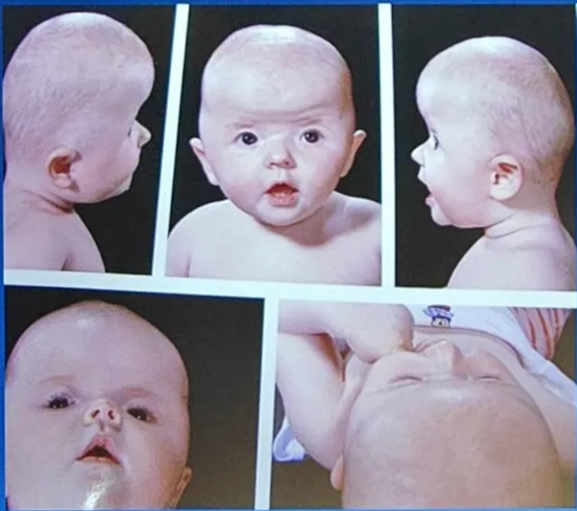
Muenke’s syndrome
- Unicoronal or bicoronal craniosynostosis
- Brachydactyly
- Thimble-like middle phalanges
- Coned epiphyses
- Carpal and tarsal fusions
- Sensorineural hearing loss
- Developmental delay
- Learning difficulties
- Seizures
- FGFR3
- Autosomal dominant.
- Caused by a point mutation in Pr0250Arg in the Ig 11-111 linker region of the FGFR3 gene on chromosome 4p16.3
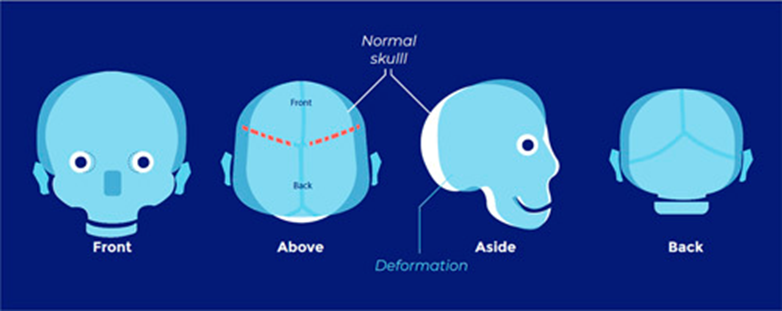
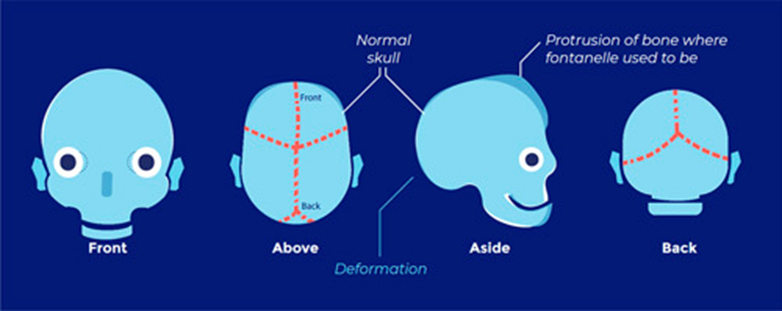
Abnormal skull shape
Low risk of raised ICP
Low risk of raised ICP
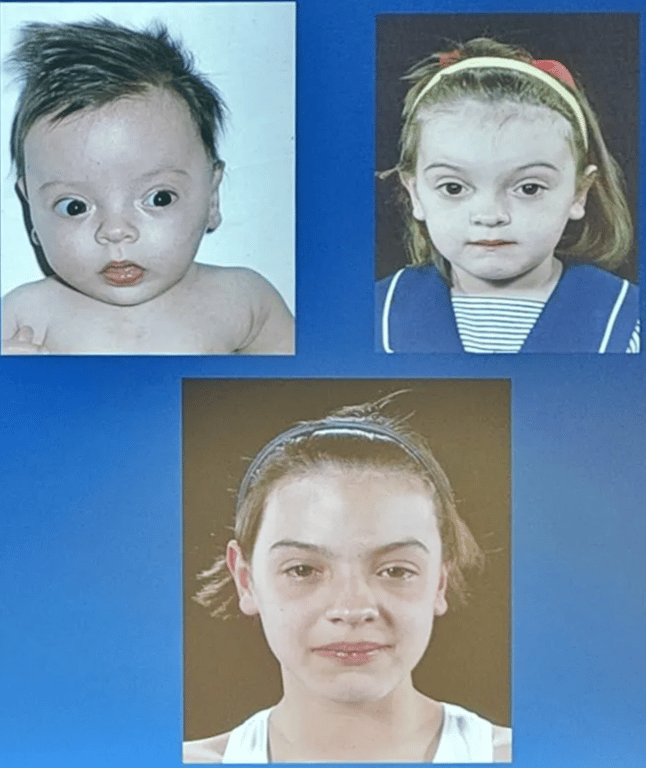
Saethre-Chotzen syndrome
- Manifestations very variable → need genetic testing
- Coronal craniosynostosis with limb abnormalities (syndactyly of the second and third digits, bifid hallux)
- Facial abnormalities (facial asymmetry, low frontal hairline, ptosis, and small ears with prominent ear crura)
- TWIST1 Chr 7
- Autosomal dominant with complete penetrance and variable expressivity.
- Caused by loss-of-function mutations in TWIST (missense, nonsense, deletions, insertions, and duplications). Report of Q289P mutation in FGFR2
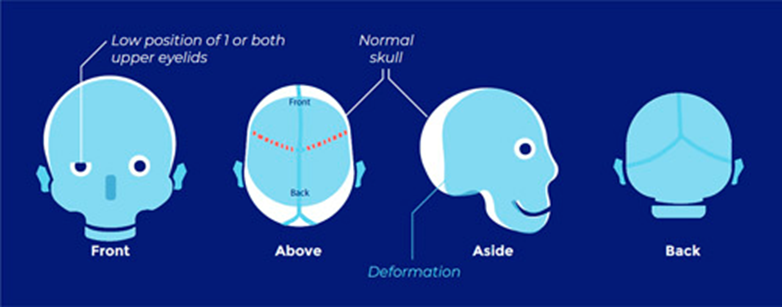
Abnormal skull shape Risk of ICP.
Crouzon’s syndrome
- Most common
- Classical triad
- Coronal synostosis
- Midfacial hypoplasia
- Exophthalmos
- Other features
- May also include involvement of other calvarial sutures
- Brachycephaly
- Hypertelorism
- Chiari I malformation
- Hydrocephalus
- Mental retardation
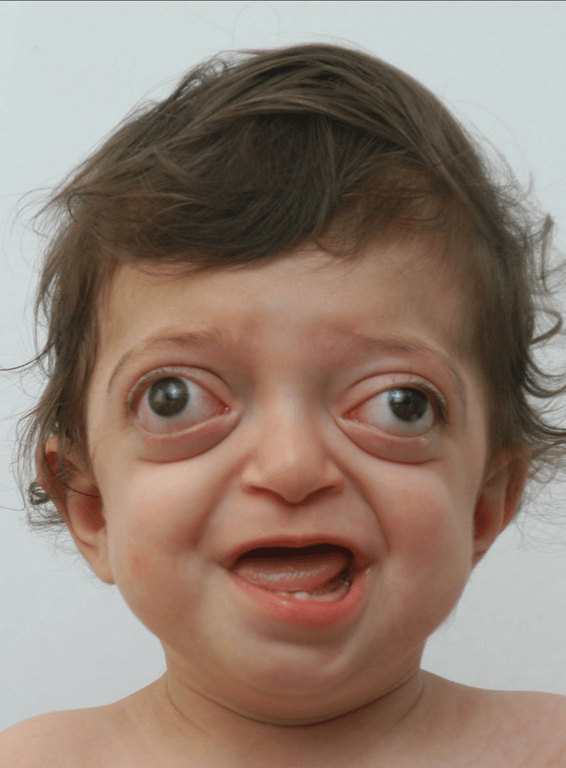
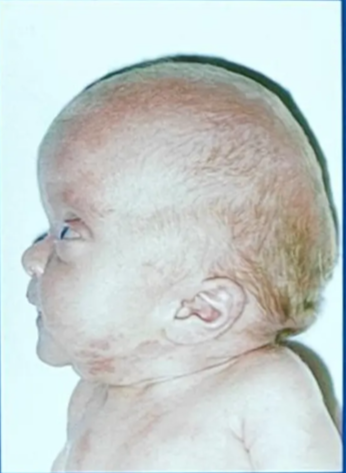
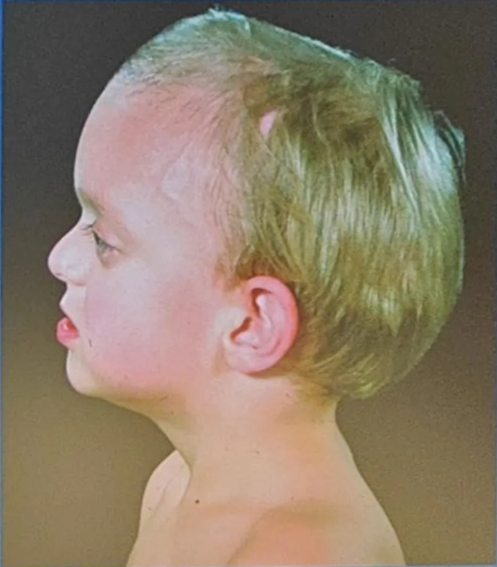
- FGFR2 Chr 10
- Autosomal dominant.
- Caused by numerous missense mutations in the Ig 111 domain of the FGFR2 gene (many involve the gain or loss of a cysteine residue)
Abnormal skull shape Risk of ICP.
Pfeiffer’s syndrome
- Classical features
- brachycephaly
- membranous syndactyly of hands and feet with enlarged and deviated thumbs and great toes
- Types
- Type I classical Pfeiffer’s syndrome is a mild entity
- Types II and III are more severe, with early death.
- Other features
- Coronal synostosis with or without premature fusion of other calvarial sutures
- Cloverleaf skull
- Facial
- maxillary hypoplasia
- small nose with a low nasal bridge
- hypertelorism
- shallow orbits
- proptosis
- strabismus
- limb malformation
- Radiohumeral synostosis
- Broad fingers and toes
- partial syndactyly of the fingers and toes
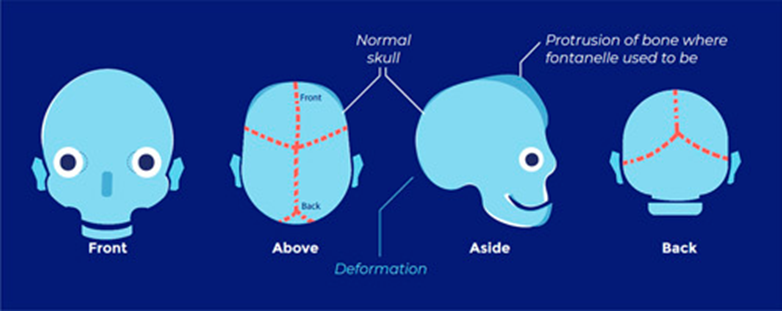

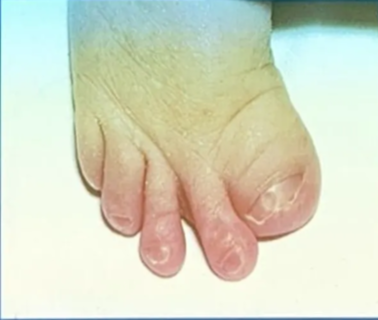
- FGFR1, FGFR2
- Type I: autosomal dominant.
- Caused by FGFR1mutations, including Pr0252Arg in the Ig II-III linker region on chromosome 8p11.2-p11.
- Can also be caused by mutations in FGFR2 and be associated with more severe phenotypic expression.
- Types II and III: sporadic inheritance.
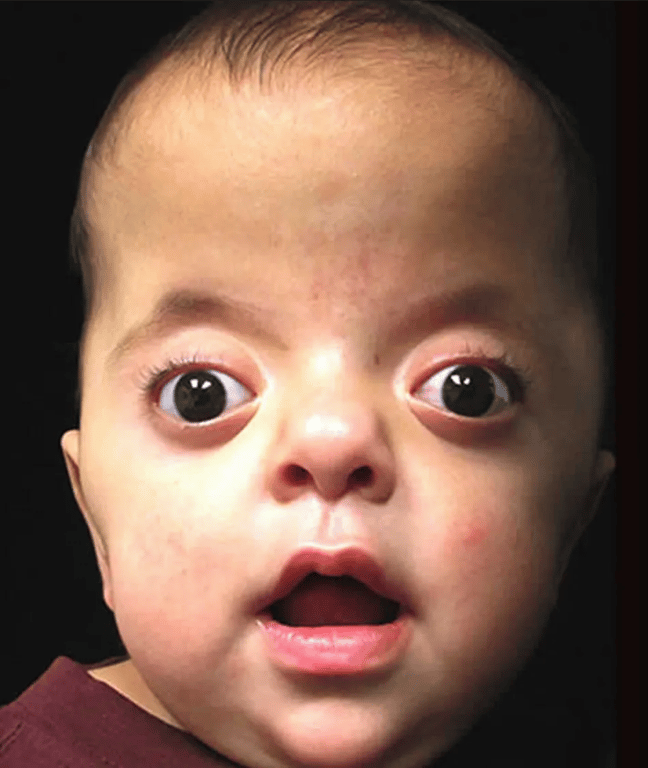
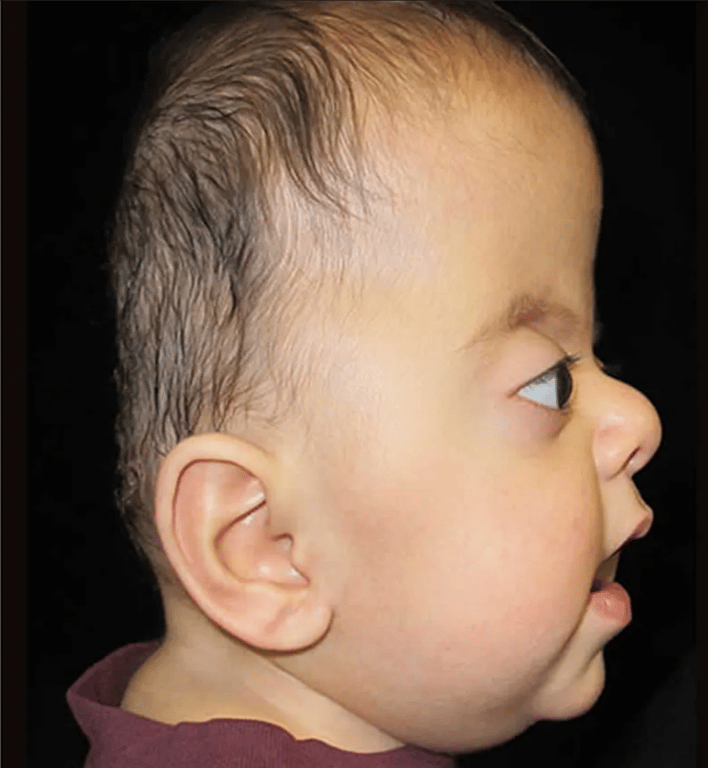
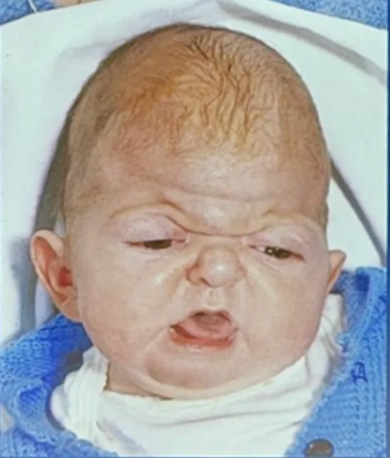
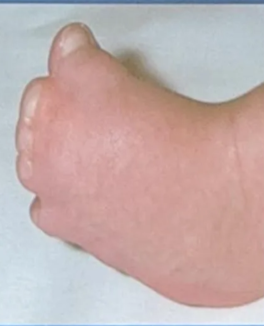
Apert’s syndrome (Acrocephalosyndactyly)
- 2nd most common
- Features
- Bicoronal synostosis
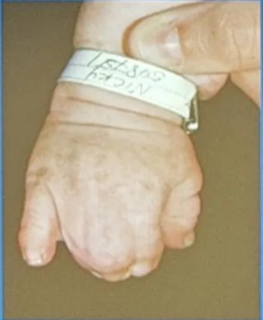
- Severe polysyndactyly in the fingers and toes
- Symphalangism (fusion of the phalanges)
- Radiohumeral fusion
- Mental retardation (IQ can be normal or mildly reduced)
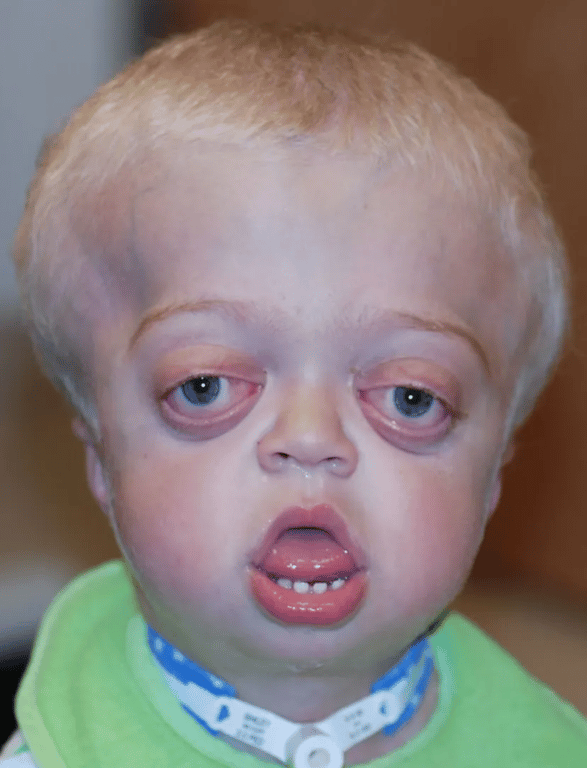
- Antimongoloid slanted eyes
- Maxillary hypoplasia
- Cheerful effect
- Vs Crouzon's at the faciocranial level
- is the presence of hypertelorism and an open bite, in which the anterior part of the maxillary alveolar arch is higher than the posterior part.
- The face and the forehead are also abnormally wide, and the anterior fontanelle is widely open during the first months of life.
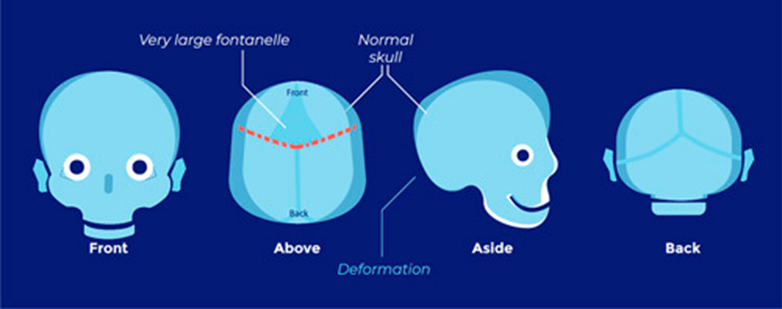
- FGFR2
- Autosomal dominant.
- Caused by a number of different mutations in FGFR2 on chromosome 10q26, including two missense mutations (Ser252Trp, Pr0253Arg) and two Alu insertions
Abnormal skull shape Risk of ICP.
Jackson-Weiss syndrome
- Like Crouzon's but with added enlarged great toes and tarsometatarsic fusion.
- Craniosynostosis, with broad toes and a medially deviated great toe, second and third toe syndactyly, tarsal-metatarsal fusion, broad and short metatarsals and proximal phalanges, midfacial hypoplasia, hypertelorism, proptosis, and normal intelligence
- FGFR2
- Autosomal dominant.
- Caused by mutation A344G in the highly conserved Ig IIIc domain of FGFR2,as well as two nucleotide missense mutations that result in Cys342Ser and Cys342Arg
 Made with Bullet
Made with Bullet